

Abstract
This paper describes the fabrication of oligonucleotide-coated Cy5-doped silica nanoparticles using a combination of multivalent linkers and their use in surface-based DNA sandwich hybridization assays. Dipodal silane is introduced as a means to fabricate amine-coated silica nanoparticles and its advantages compared to monopodal silanes are discussed. The use of dipodal silane in conjunction with three different polymer linkers (oxidized dextran, linear and 8-arm polyethylene glycol (PEG)) to immobilize single-stranded DNA to Cy5-doped nanoparticles is investigated and dynamic light scattering measurements and Fourier transform infrared spectroscopy are used to follow the progression of the functionalization of the nanoparticles. We observe a significant improvement in the binding stability of the single-stranded DNA when the dipodal silane and 8-arm PEG are used in combination, when compared to alternative conjugation strategies. Both 8mer and 22mer oligonucleotides are securely conjugated to the high-brightness nanoparticles and their availability to hybridize with a complementary strand is confirmed using solution-based DNA hybridization experiments. In addition, a full surface-based sandwich assay demonstrates the potential these nanoparticles have in the detection of less than 500 femtomolar of a DNA analogue of micro RNA, miR-451.
Export citation and abstract BibTeX RIS
Introduction
Highly fluorescent dye-doped silica nanoparticles (NPs) have great potential as labels in biomedical diagnostics such as for the detection of nucleic acid or protein biomarkers. Silica NPs are optically transparent and can be modified using a wide range of organosilane linkers for bioconjugation [1–3]. In addition, silica NPs are biocompatible [4, 5], photostable [6] and protect fluorescent dyes from molecular quenchers. A wide range of fluorescent dyes can be loaded inside silica NPs for high sensitivity detection of biomarkers or multiplexing applications using either the reverse micro-emulsion or Stöber method [7–11].
For point-of-care (POC) devices, dye-doped silica NPs are attracting significant attention and have already been integrated into commercial products [12–15]. With a large amount of dye encapsulation, the increased brightness of these NPs results in improved signal-to-noise ratios compared to free dye labels [8]. Furthermore, synthesis can be achieved using inexpensive alkoxide precursors at room temperature [16]. When developing POC assays, the binding of the active biomolecule (which interacts directly with the target) to the surface of the high-brightness nanoparticle is also favourable as it limits the number of steps required to run the full assay. Another important factor that must be taken into account is that the biomolecules e.g. oligonucleotides, antibodies and enzymes, are bound in a stable manner to the nanoparticle surface; for this, covalent immobilization is preferable to physisorption [17]. However, it is vital that with such immobilization the biomolecules not only remain attached under vigorous washing in aqueous conditions, but that they also remain active and available for binding.
When choosing immobilization strategies for linking of biomolecules to silica nanoparticles, the use of silanes for surface activation is commonplace. In particular, aminopropyltriethoxysilane (APTES) linkers are used to provide the surface of the silica with amine groups, to which biomolecules can be directly linked. However, there is evidence that these APTES groups can become chemically unstable at high and low pH, and at raised temperatures, conditions often experienced during assay development [18–20]. In this work, we investigated the use of the dipodal silane, bis [3-(triethoxysilyl) propyl] amine, (ABisTES), which increases crosslinking density of silanes on silica surfaces i.e. ABisTES is able to form six bonds to the NP per silane compared to three for APTES (see figure 1) [21]. The principal of improving the stability of binding of linkers by increasing the number of attachment points has been previously demonstrated with multidentate thiol ligands for attaching oligonucleotides to gold nanoparticles [22–25]. Dipodal silanes participate in covalent binding with silica surfaces and have previously been recommended in the fields of catalysis and separation science when such surfaces are used under aqueous conditions, where the hydrolytic stability of the siloxane bond between the surface and functionalization group is unstable [19, 20]. We hypothesize that the use of ABisTES to provide the surface of the silica nanoparticle with amine functionalisation would improve the chemical stability of these groups on the surface, by limiting the rate at which hydrolysis occurred. We are not aware of previous work on the use of dipodal silanes to functionalize silica nanoparticles.

Figure 1. Schematic showing the conjugation approach for nanoparticle functionalization using activation silanes, ABisTES and APTES, and linkers, linear PEG, 8-arm PEG and oxidized dextran.
Download figure:
Standard image High-resolution imageThe binding of biomolecules directly to surfaces can have a negative effect on the availability of the biomolecules to interact with target molecules due to restricted movement and steric hinderance. In these instances, the use of spacers and linkers provide may not only improved binding stability but also improve the availablity of bound biomolecules for interaction with the required targets. In addition, selection of the correct linkers can provide nanoparticles with increased colloidal stability [17, 26, 27]. Multivalent linkers have shown to be highly effective for stable binding of biomolecules to surfaces: in previous work using multivalent dendrimer polymer linkers for application in immunoassay, significant improvements in antibody binding and assay performance were observed [28]. Multivalent polymer linkers have also improved binding and availability of oligomers on surfaces [29–32].
We are currently developing nucleic-acid conjugated, dye-doped silica NPs for use in POC diagnostic systems for the detection of micro RNA biomarkers. When working with silica NPs where APTES was used to provide the amine surface and small, linear linkers to immobilize oligonucleotide probes, we observed substantial loss of the oligonucleotides from the surface after washing (see figure S1 in supplementary information). This led us to investigate the method of using dipodal silanes combined with multivalent linkers multi-arm polyethylene glycol (8PEG) and oxidized dextran (OxDex) to immobilize the oligonucleotides. Linear polyethylene glycol (LPEG) was investigated as a linear comparison. Figure 1 outlines the stages at which the different silanes and linkers were employed. These linkers were chosen to provide improved availability of the bound oligonucleotides, as well as possible reduction in non-specific binding (NSB), a factor crucial to control when developing surface-based assays [33, 34]. 8PEG has previously been used in the formation of polymer networks for tissue engineering and in nanoaggregate synthesis but it has not been utilized as a functional coating for NPs until now [35–37]. OxDex has been used previously as a coating for lab-on-a-chip devices to immobilize antibodies and for the immobilization of streptavidin to silica NPs, and dextran has been used previously to coat NPs for calcium sensing applications [38–40].
The key features of this paper include (i) the novel use of a dipodal silane to improve functionality of silica nanoparticles, (ii) use of 8-arm PEG linker for greater oligonucleotide surface loading and (iii) use of these nanoparticles to detect of a DNA analogue of the micro RNA, miR-451 at femtomolar concentrations in a surface-based hybridization assay.
Materials and methods
Materials
Water soluble, Cy5 mono-reactive NHS ester was purchased from Lumiprobe (USA). Anhydrous n-hexanol, 3-aminopropyltrimethoxysilane (APTMS, 99%), 3-aminopropyltriethoxysilane (APTES, 99%), bis [3-(triethoxysilyl) propyl] amine (95%) (dipodal silane), 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol, t-octylphenoxypolyethoxyethanol polyethylene glycol tert-octylphenyl ether (Triton® X-100), polyoxyethylene (5) nonylphenylether (NP5), dioctyl sulfosuccinate, sodium salt (AOT, 98%), cyclohexane (anhydrous, 99.5%), n-hexane (anhydrous, 95%), tetraethylorthosilicate (TEOS, 99.999%), phosphate buffered saline tablets (one tablet dissolved in 200 mL of deionised water yields 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride, pH 7.4, at 25°C), disodium phosphate (>99%), monosodium phosphate (>99%), hydrochloric acid (1.0 N), 2-(N-morpholino)ethanesulfonic acid hydrate (MES hydrate, >99.5%), ammonium hydroxide solution (28% w/w in water), N-hydroxysuccinimide (NHS) (98%), O,O'-bis[2-(N-succinimidyl-succinylamino)ethyl] polyethylene glycol (LPEG-NHS) (2000 Da), sodium periodinate (ACS reagent, ≥99.8%), diglycolic anhydride (90%), potassium bromide (≥99%), phosphoric acid (ACS reagent, ≥85 wt% in H2O), glutaraldehyde (50% wt in water), sodium borohydride (98%), Micro90, and albumin from bovine serum (BSA, freeze dried powder, ≥96%) were all purchased from Sigma Aldrich, Dublin, Ireland. Deionized water (<18 M Ω) was obtained from a MilliQ system from Millipore, Ireland. 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC) (98+%) was purchased from Acros Organics, Geel, Belgium. 8-arm PEG succinimidyl NHS ester (8PEG-NHS) (10 000 Da) was purchased from Nanocs Inc. New York, USA. Glass vials were purchased from Chemglass. Sterile/DNAase-free tips using TipOne® technology were purchased from StarLabs. Oligonucleotides were purchased from Eurofins MWG Operon (D-85560 Ebersberg, Germany).
Instrumentation
Fluorescence measurements were performed on a Safire (Tecan) microplate reader. For Cy5-doped NPs, the excitation and emission wavelengths were set at 646 and 676 nm, respectively and for the Cy3-labelled oligonucleotide, the excitation and emission wavelengths were set at 546 and 576 nm, respectively. To determine the exact concentration of Cy5 and Cy3 on the nanoparticles, calibration curves of free dye in water were calculated from a cascade dilution (see figure S2 in supplementary information). Surface-based assays were imaged using a microarray scanner (Perkin Elmer Scan Array GX). Microarrays were fabricated by non-contact spotting (Scienion S3 arrayer), equipped with a PDC-90 nozzle.
Particle sizes and zeta potentials of samples in solution phase were determined using dynamic and electrophoretic light scattering analysis performed on a Delsa™ Nano C from Beckman Coulter. Each measurement was performed using the same concentration (0.5 mg mL−1) and pH (7.0) to minimize errors from changing ionic strength, NP concentration or pH. The standard deviation of the zeta potential was found to be less than 14% from multiple replicates performed on test samples. With regard to size calculations, curve fitting programs with the Delsa™ Nano C software were used to calculate the average NP diameter and standard deviations from samples where the polydispersity index was lower than 0.3.
Solid-state, diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out in duplicate using a Perkin-Elmer GX FT-IR in order to calculate the quantity of amine groups on the surface of each NP. Dried NP samples of equal weight (3% w/w) were mixed with a stock sample of ground KBr with a constant scattering coefficient [41]. A Kubelka–Munk conversion was performed on background-subtracted samples to obtain quantitative results. The Kubelka–Munk equation (1), where R is the absolute reflectance of the sampled layer, k is the molar absorption coefficient and s is the scattering coefficient, creates a linear relationship for spectral intensity relative to sample concentration and is ideal for powdered samples [42]
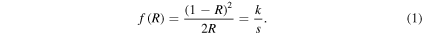
Methods
Functionalization of silica NPs with monopodal/ dipodal silanes
Cy5-doped silica NPs were synthesized using an optimized protocol from our previous paper using the microemulsion method [9]. In this work, NPs were also coated with a thin silica shell using the Stöber method [43]. A significantly greater performance was achieved using this approach because addition of a pure silica shell creates a surface layer of reactive silanol groups separated from the dye-doped region [44]. Silica NPs, as-prepared and after addition of the shell, were highly monodispersed with average diameters of 42±1 nm and 49 ± 2 nm, respectively, (see figure S3(a) and figure S3(b) in the supplementary information). For silane attachment, NPs (6 mg) previously suspended in ethanol (950 μL) were mixed with MilliQ-H2O (50 μL). Following this, 3-aminopropylethoxysilane (APTES) (for monopodal silane) or bis [3-(triethoxysilyl) propyl] amine (for dipodal silane) (20 μL) was added and the reaction was stirred at room temperature for three hours. The reaction time of three hours was chosen for the amine functionalization as Roy et al observed the formation of a monolayer of APTES on silica NPs after this time [44]. The NPs were then washed and centrifuged three times in ethanol (17 000 × g, 10 min). Finally, the NPs were conditioned in phosphoric acid solution (pH 7, 0.01 M) for 16 h at room temperature to remove any non-specifically bound silane.
Conjugation of linkers to NPs
LPEG and 8PEG were conjugated to the amine surface via amide bond formation between an amine (on NP surface) and an N-hydrosuccinimde (NHS) ester on the PEG; amines readily reacted with NHS esters to form a covalent amide bond. However, it was found that during conjugation, the addition of 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was necessary to ensure efficient binding, as NHS esters are known to hydrolyze in the presence of water. In the case of OxDex, the linker was attached via a Schiff base reaction of the aldehyde on the OxDex and the amine on the surface. Based on previously reported work, a time of 2 h was used when oxidizing the dextran, prior to reacting with the nanoparticles [38]. This reaction time results in an approximate value of 40% conversion from alcohol to aldehyde [45].
PEG-linkers : Amine-coated NPs (∅ = 50 ± 1 nm, 2 mg) were suspended into 1 ml of MES buffer (pH 4.7, 0.1 M, previously filtered). To this was added LPEG/8PEG-NHS (1 mg) dissolved in 500 μL of MES buffer to a final concentration of 50 mM EDC. This sample was reacted for 2 h at room temperature and washed twice in ethanol for purification (3 centrifugations, 17 000 × g, 7 min). OxDex-linker : Amine-coated NPs (∅ = 50 ± 1 nm, 2 mg) were suspended into an OxDex solution (1% w/v) in PBS buffer (1 mL, pH 4.7, 0.01 M). The sample was stirred for 2 h at room temperature, washed in MilliQ-H2O and purified in ethanol (3 centrifugations, 17 000 × g, 7 min). Anhydride linker : APTES-coated NPs (1 mg) were suspended into 0.5 mL of phosphoric acid buffer (0.5 M, pH 7.0). To this was added diglycolic anhydride (6.6 mg) previously dissolved in 0.5 mL of the same solution. The solution was stirred for 16 h at room temperature and washed twice in ethanol (3 centrifugations, 17 000 × g, 7 min).
Conjugation of ssDNACy3 to linker-coated NPs
LPEGNP and 8PEGNP (200 μL, 1 mg mL−1, 9.4 nM, in MES, pH 4.7, 0.1 M) were mixed with amine-terminated, Cy3-labelled oligonucleotides (table 1) (10 μL, 100 μM, aliquots in freezer). This was made up to 500 μL with the addition of EDC (to a final EDC concentration of 50 mM). OxDexNP (200 μL, 1 mg mL−1, 9.4 nM, in PBS, pH 7.0, 0.01 M) were mixed with amine-terminated, Cy3-labelled oligonucleotides (10 μL, 100 μM, aliquots in freezer). All samples were reacted for 2 h with shaking at room temperature in the dark. The samples were then extensively washed with nuclease-free water, and stored in 500 μL of nuclease-free water for measurement.
Table 1. ssDNA oligomer sequences used; Cy3 = Cyanine 3, Cy5 = Cyanine 5, and Dabcyl = 4-(4'-dimethylaminophenylazo) benzoic acid.
| Entry | Structure | Reference name |
|---|---|---|
| a |

|
22mer ssDNACy3 |
| b |

|
8mer ssDNACy3 |
| c |

|
3' dabcyl complementary target |
| d |

|
Internal dabcyl complementary target |
| e |

|
Surface assay capture probe |
| f |
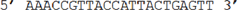
|
Surface assay target |
| g |

|
Surface assay reporter probe (bound to NP) |
| h |

|
Surface assay reporter probe (free dye) |
Fluorescence spectroscopy
NPs were coated with ssDNACy3 and the ratio of the fluorescence signal from the Cy5 (NP) and Cy3 (ssDNA) dyes enables the number of oligonucleotides conjugated to the surface of the NP to be calculated as these two dyes have different excitation and emission wavelengths. For these experiments, two references were used: (1) a solution of NPs of known concentration (with no conjugated oligonucleotides) and (2) varying concentrations of ssDNACy3 in nuclease free water which were used to generate a calibration curve The absorption and fluorescence of the Cy5-doped NPs (without oligonucleotides) at 1 mg mL−1 were also determined using the Tecan scanner.
Robustness of oligonucleotide binding to NPs: washing protocol
To determine the stability of the oligonucleotide conjugation (via the amine-functionalization and linking steps), washing experiments were carried out in duplicate. ssDNACy3–8PEGNP/−LPEGNP/OxDexNP (0.8 mg) were suspended in 2X-sodium saline citrate (SSC) buffer (2 mL) containing sodium dodecyl sulfate (SDS) (pH 7.0, 0.01%), hereafter known as 'SDS buffer'. The fluorescence intensity was measured to obtain a starting fluorescence measurement. Following a brief ageing time (5 min) samples were spun down and 200 μL of supernatant collected from each aliquot. The NPs were then resuspended into 2 mL of SDS buffer and 200 μL of the resuspended NP solution collected. The fluorescence of both supernatant samples and resuspended NP samples was then measured. Following a further short ageing time, the remaining 1800 μL resuspended NP samples were centrifuged and again 200 μL of supernatant was collected from each aliquot. The NP samples were then resuspended into 1800 μL of buffer and a further 200 μL of NP solution collected. The fluorescence of the second set of supernatant samples and resuspended NP samples was then measured. Further cycles were repeated until the standard deviation from repeat measurements was less than 5%. The ratio of the Cy3 to Cy5 fluorescence signals is directly proportional to the number of Cy3-labelled oligonucleotide attached to the NP as the fluorescence signal related to the emission wavelength of the Cy3 dye comes from the oligonucleotide bound to the Cy5-doped NP. The number of oligomers was calculated using the concentration of both the Cy5 dye (CCy5) and the Cy3 dye (CCy3) present in the NP samples, using equations (2) and (3) below, where FI676 is the fluorescence intensity of the samples measured at the emission wavelength of Cy5 (676 nm), and FI563 is the fluorescence intensity of the samples measured at the emission wavelength of Cy3 (576 nm). Two calibration curves of the change in fluorescence relative to concentration for pure Cy5 NPs and Cy3 free dye were determined from a cascade dilution (see figure S2 in the supplementary information). The number of NPs in each sample was determined our previously published method [9]. Adaptation of previously published methods enables us to calculate the number of oligonucleotides per nanoparticle using the equations below;
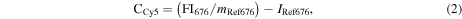
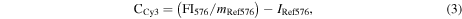
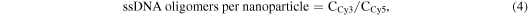
where mRef662 and mRef563 are the gradients and IRef662 and IRef563 are the intercepts determined from the respective calibration curves. The number of DNA strands per nm2 was calculated by first calculating the number of oligomers per nanoparticle, followed by calculation of the average surface area of a nanoparticle (surface area of a sphere = 4πr2 (r = radius in cm). The number of oligomers per nanoparticle was divided by the total area of the nanoparticle surface to calculate the area occupied for each strand on average.
Solution-based oligonucleotide hybridization on NPs: washing protocol
ssDNACy3–8PEGNP (0.4 mg, 9.4 nM) were suspended in SDS buffer (pH 7.0). Previously, the NPs were washed three times to reach a stable number of oligonucleotides on the surface. To this was added 20 μL of the complementary ssDNA (100 μM). For direct-binding hybridization, the NPs conjugated to the 22mer ssDNACy3 and complementary oligomers with a dabcyl quencher at the 3' end were used. For the sandwich hybridization, the NPs conjugated to the 8mer ssDNACy3 and a complementary oligomer containing an internal dabcyl quencher was used (a schematic of both hybridization reactions is shown later). The samples were then reacted for 2 h with mixing at room temperature in the dark. The NPs were then washed 5 times, after which the fluorescence of the samples was measured again. Upon hybridization of the dabcyl-containing oligomers, the Cy3-signal on the complementary oligomers would be reduced, thus indicating whether the oligonucleotides conjugated to the NP was available for successful hybridization. The exact number of dye-labelled oligonucleotides that were quenched was determined using the linear equations and calibration curves described previously.
Preparation of slides for surface assay
Glass slides were functionalized with amine groups in order to bind amine-terminated oligonucleotide capture probes using the following method: glass microscope slides were cleaned by submersing in 2% micro90 and sonicated for 30 min. They were then rinsed with deionized water, submersed in ethanol and sonicated for a further 30 min. Post cleaning, slides were dried in nitrogen and placed in an oxygen plasma for 5–10 min. Immediately after plasma activation, the slides were submersed in a 3% w/w APTES solution in 95% ethanol for 2 h. Slides were then sonicated in ethanol for 15 min (×2) and finally rinsed with ethanol and dried under nitrogen. APTES-coated slides were then baked in an oven at 70°C for 1 h.
Slides were then activated for reaction with the amine-terminated oligonucleotide and blocked to reduce any possible NSB and deactivating the glutaraldehyde linker: slides were submersed in 1% glutaraldehyde solution in phosphoric acid (pH 7.0) for 1 h at room temperature. Immediately after, slides were dried in nitrogen and spotted with amine-terminated DNA capture probes (Entry e, table 1). Spotting was carried out by suspending oligonucleotides in 3X SSC buffer at a concentration of 10 μM and depositing them onto the surface of the activated APTES-coated slides using a Scienion piezoelectric spotter equipped with a PDC-90 nozzle, after which they were left in ambient conditions at room temperature overnight. Each microspot was generated from 2 drops of the spotter. Blocking with BSA was then carried out by submersing slides in a 3% BSA solution (pH 7.0) for 1 h at room temperature. After these slides were added to the aldehyde blocking solution (0.25 g of NaBH4 in 75 mls of PBS and 25 mls of ethanol prepared 30 min ahead of use) for 20 min followed by a 2 × 2 min wash with 0.2X SSC and 2 min of deionized water. The slides were then dried by centrifuge.
DNA hybridization assay protocol
Mixtures of target DNA (Entry f, table 1) (40 pM–0 pM) and reporter solutions were made up to 200 μL in hybridization buffer (2x SSC, 0.1% SDS and 1% BSA, pH 7.0). In the case of the free dye, (Entry h, table 1) reporter solution at a concentration of 200 μM was used. For the reporter probe-coated NPs a concentration of 0.035 mg mL−1 of particles was used. These target/reporter mixtures were then heated at 70 °C for 3 min and further incubated at 30 °C for 30 min before being added to the slides containing the capture probe and incubated overnight at 30 °C. After hybridization, solutions were removed from the slide and the wells were washed (2X SSC and 0.2% SDS for 10 min at 30 °C, with 2X SSC for 10 min at room temperature and with 0.2X SSC for 10 min at room temperature for free dye; twice with PBS and once with deionised water for Cy5-nanoparticles). The slides were finally dried by centrifugation and imaged using the Perkin Elmer fluorescent scanner. All measurements were carried out in triplicate.
Results and discussion
Dipodal silane versus monopodal organosilane
After activation of silica nanoparticles with dipodal silane, ABisTES, we compared the properties of the particles to those activated with monopodal silane, APTES. This was carried out using Fourier transform infrared (FTIR) spectroscopy and dynamic light scattering (DLS). Diffuse reflectance FTIR analysis confirmed the presence of the amines on both the APTES and ABisTES samples. The characteristic bands for the N–H stretching and bending vibrations were found at 3300–3400 cm−1 and 1620–1650 cm−1 respectively. However, these bands overlapped with the stretching and bending vibrations of the Si–OH groups in the silica NP and so they themselves could not be used as reference for this experiment. For that reason, the presence of asymmetric and symmetric stretching vibrations of the CH2 band on the propyl chain was followed as an indication of the presence of the silane (between 2800 and 3000 cm−1) [19]. After each measurement, a Kubelka–Monk conversion was carried out to obtain quantitative results (figure 2).
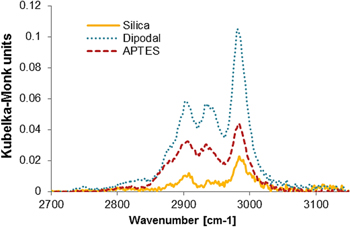
Figure 2. Kubelka–Monk conversion DRIFTS measurements of dipodal-coated, APTES-coated and pure silica nanoparticles.
Download figure:
Standard image High-resolution imageIt was observed that the use of ABisTES resulted in slightly greater than double the signal obtained from the APTES coating. This was not unexpected since ABisTES has twice the number of CH2 groups when compared to APTES. For pure silica NPs the C–H stretching vibrations observed here are due to either incomplete alkoxide hydrolysis or residual ethanol from the drying process. In summary, both aminosilanes bound to silica NPs but ABisTES was slightly more efficient.
For both amine-coated NPs, zeta potential measurements also gave an indication of the stability of the groups on the surface, with highly amine-functionalized surfaces having a strong positive charge and amine free surfaces showing the negative charge of deprotonated silanol groups on the silica surface. The zeta potential of amine-coated NPs in phosphoric acid at 24 h intervals after synthesis showed that APTES and ABisTES particles both remained positive (>20 mV) up to 168 h in solution; demonstrating that the amine groups were still present on both samples and that hydrolysis was not occurring on these samples under these conditions at neutral pH. However, upon purification and resuspension in phosphoric acid solution, ABisTES-coated NPs were colloidally stable whereas the APTES-coated NPs were observed to aggregate in less than two minutes (see figure S4 in supplementary information). A similar improvement in colloidal stability was also observed when bidentate thiol ligands were attached to gold nanoparticles; however, the thiol-gold bond is reversible and the use of this bidentate ligand did not lead to improved bioconjugation [46]. The size of the ABisTES-coated NPs was determined after 5 and 30 min using DLS and was found to be 106 ± 28.4 nm and 84.9 ± 25.1 nm respectively. It was not possible to measure the size of the APTES-coated NPs due to rapid aggregation. In summary, the DLS analysis demonstrated that the amine functionalization for both the APTES and ABisTES NPs was stable in phosphoric acid and that significant hydrolysis of the conjugated silanes was not observed. Although the reason for investigating ABisTES as an alternative to APTES was primarily to investigate hydrolytic stability, it was this significant improvement in colloidal stability that led us to recommend ABisTES linkers for surface activation as a first step in silica bioconjugation.
Linker optimization
We proceeded to use ABisTES-coated nanoparticles in conjunction with multivalent linkers to attempt to improve the binding of the oligonucleotides. We selected OxDex and eight-arm polyethylene glycol (8PEG) as multivalent linkers, and LPEG as a monovalent comparison. After attachment to the NP as described in the materials and methods section, the OxDexNP, LPEGNP and 8PEGNP were then conjugated to amine-terminated, a Cy3-labelled oligonucleotide (Entries a, b or g in table 1, depending on the application). Figure 3 shows the Cy3 fluorescence signal from nanoparticles coated with 22mer ssDNACy3 (Entry a, table 1) bound via the three different linkers, initially and after three washes in SDS buffer. The rapid aggregation of the APTES particles described above could also be the reason for the poor conjugation of the DNA observed in our initial experiments as we did not observe to have the same difficulty in achieving secure binding using ABisTES.
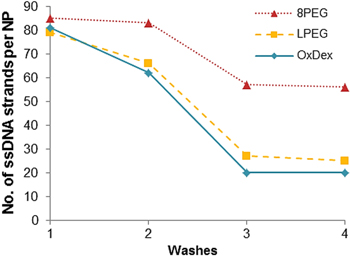
Figure 3. Number of DNA strands calculated, using fluorescent measurements, to be bound to the ABisTES-activated high-brightness NPs via different linkers (8-arm PEG, linear PEG and oxidized dextran, initially and after washing in SDS buffer. Washing in SDS removes the non-specifically bound Cy3-labelled ssDNA from the surface.
Download figure:
Standard image High-resolution imageAll three samples bound a large amount of DNACy3 (79 strands for LPEGNP, 81 strands for OxDexNP and 85 for 8PEGNP) and a strong fluorescence signal was seen. The first washing step in SDS buffer reduced the amount of ssDNA bound to the surface of each sample (66 strands for LPEGNP, 62 strands for OxDexNP and 83 for 8PEGNP). It was assumed that during this wash step most of the non-covalently bound ssDNA was removed. Further washes show that the DNA binding stabilizes after three washes, with 57 strands per NP calculated for the 8PEGNP indicating that it provided the strongest binding of all linkers. Combined with the ABisTES functionalization, 8PEGNP not only initially bound more oligonucleotides than the OxDexNP or LPEGNP samples but also and more importantly, the NP was shown to retain the largest amount of ssDNA strands (approximately twice as much as OxDex or LPEG linked particles) after successive washing. We believe that the multivalency of the 8PEG linker allowed for multiple covalent bonds to be formed between the 8PEG and the amine coating on the NP as well as multiple ssDNA strands to bind to the 8PEG. This improved binding of the oligonucleotides agreed with our previous work on the use of multivalent linkers to attach antibodies to NP labels for use in immunoassays, where the use of multivalent linkers led to a higher efficiency in bioconjugation of detection antibodies [28, 47]. In the case of the LPEG linker, there was only one binding point available for attachment to the dipodal silane, which could have been more readily available for hydrolysis than binding points on a multivalent linker, resulting in fewer ssDNA strands be attached to the surface via covalent bonding. In the case of the OxDex linker, initial oligonucleotide binding was strong but levelled out after 2 washes to a value comparable to that of LPEG. We postulate that the decreased number of ssDNA conjugated to OxDex compared to the multivalent 8PEG was due to fewer functional groups available for binding, as approximately 40% of the alcohol groups on OxDex were converted to aldehydes during the 2 h oxidization reaction [45]. Some loss of functionality of the OxDex may also be attributed to the hydrolysis of the enamine group formed during the Schiff-base reaction in the SDS buffer. It was therefore concluded that the optimum conjugation combination was the dipodal functionalisation with the 8PEG linker (8PEGNP-DNA).
A number of control experiments were carried out in order to ensure that the binding of the oligomers was covalent in nature. The zeta potential of 8PEGNP in different buffers (pH 7.0, or in the case of MES, pH 4.7) was calculated at room temperature and the results (figure 4) confirm that the nanoparticles are generally either neutral or negatively charged, making the physical adsorption of negatively charged oligonucleotides highly unlikely. Further analysis of the stability of these silica nanoparticles can be found in other published work by this group [48]. Experiments were also performed without the addition of EDC to activate linkers during the conjugation steps and the binding of the oligonucleotide was not successful (see figure S5 in the supplementary information), demonstrating that the binding is indeed covalent and not merely physical. Finally, the robust binding of amine-terminated oligomers to the surface was demonstrated using a probe control where there was no terminal amine group. Figure S5 (supplementary information) also shows the lack of stable binding of this non-modified oligonucleotide to the 8PEGNP in the presence of EDC activation. This confirms that little or no physical adsorption of oligomers takes place on the surface of the 8PEGNP.
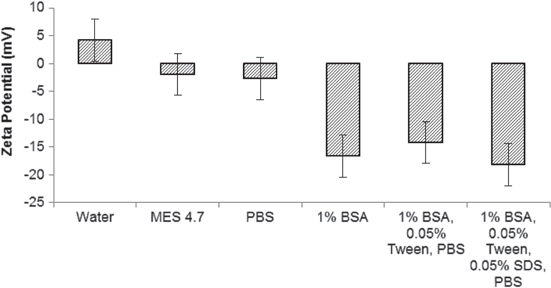
Figure 4. Zeta potential of 8PEG-coated nanoparticles in different buffers as measured by DLS. Error bars represent the standard deviations of calculated from the DeltaNano software.
Download figure:
Standard image High-resolution imageIn previous work by Delport et al, amine-functionalised ssDNA with an ATTO 647N flurophore label were coupled to silica NPs (∅ = 249 nm) using carbodiimide chemistry, in which 80 ssDNA oligomers were shown to attach per NP [49]. This corresponds to approximately one ssDNA strand per 2430 nm2 of silica surface. Using the ABisTES-8PEG system, we found that approximately 16 ssDNA are immobilized per unit area compared to 1 ssDNA strand using direct linking to a carboxyl functionalised surface (see materials and methods section for procedure). TEM images of the 8PEGNP are also shown in figure S3d (supplementary information) and the mean diameter of the particles was calculated to be 53 ± 2 nm. Using carboxyl activated surface and direct linking, we attached four ssDNA strands per silica NP (∅ = 80 nm), corresponding to approximately 1 ssDNA strand per 5000 nm2 (data not shown); again this was significantly lower than that obtained using this multivalent approach. When calculating the number of DNA strands on the surface of the nanoparticle, we were careful to ensure that there was no quenching of signals from any Förster resonance energy transfer between the Cy3 on the surface and the Cy5 in the nanoparticle (see figure S6 in the supplementary information).
Availability of conjugated DNA
It was important to ensure that the full oligomer chain bound to the NP was available for hybridization with its complementary strand and not sterically hindered by the hydrophilic, porous polymer coating. To demonstrate this, fluorescence quenching studies from hybridization with complementary DNA containing dabcyl quenchers were carried out. We performed these experiments on both ABisTES and APTES functionalized 8PEG-coated nanoparticles; this enabled us to directly compare the use of 8PEG with both silane linkers. Figure 5(a) shows a schematic of the two single-stranded reporter probes bound to the 8PEG-coated NP; one with a 22mer DNACy3 and another with an 8mer DNACy3 (Entries a and b, table 1). The complementary targets containing the dabcyl quencher were then added to this solution. For the 22-mer reporter probe, the target contained a dabcyl moiety at the 3' end, and for the 8-mer reporter probe, an internal dabcyl was used (Entries c and d, table 1). After the complementary targets were introduced, the washing of the NPs (five times) ensured that any non-specifically bound target was removed. Figure 5(b) presents the hybridization availability as a percentage of probes covalently bound to each nanoparticle.

Figure 5. (a) ssDNA targets and probes used to test hybridization. Upon hybridization of the complementary strand, dabcyl quencher will reduce the fluorescence signal from the Cy3-labelled conjugated ssDNA; (b) fluorescent signal from Cy3-labelled 22mer and 8mer probes on ABisTES and APTES 8PEG NPs which have been hybridized with quencher probes. Graph shows the percentage DNA strands which were hybridized (fluorescent signal was quenched) and which were not (fluorescent signal remained unquenched).
Download figure:
Standard image High-resolution imageFor both the ABisTES and the APTES particles, the quenching effect can be seen by the considerable reduction in the signal from the dye-labelled oligonucleotides (88% loss of signal from the 22mer and 69% loss of signal from the 8mer for the ABisTES, and 79% loss of signal from the 22mer and 60% loss of signal from the 8mer for the APTES). The high negative charge of both the 8PEG-coated NPs and the oligonucleotide-coated NPs combined with a large amount of washing ensured that the target strand did not bind non-specifically. Knowing that the ssDNA is bound in a robust manner to the NP, we attribute the large reduction in fluorescence to the dabcyl quencher being in very close proximity to the Cy3 dye. We believe that the target oligonucleotide is participating in full hybridization with both strands bound to the silica NP and that therefore, the ssDNA bound to the surface was available for hybridization for either direct binding or sandwich binding, and that steric interference or entrapment from the polymer coating did not affect the binding of the complementary strand. We also attribute the stability of the DNA strands on the APTES-coated particles to the use of the 8PEG linker.
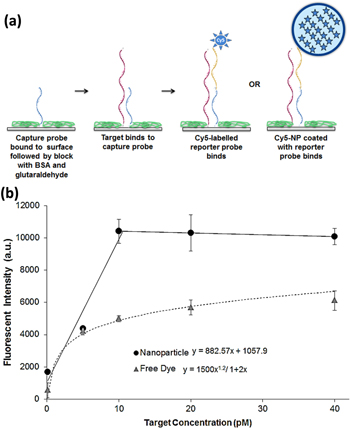
To exemplify the potential applicability of our assay, we applied it to the detection of a DNA analogue of the micro RNA, miR-451. miR-451 is widely dysregulated in human cancers and plays a critical role in tumourigenesis and tumour progression [50]. miR-451 is present in human plasma at picomolar concentrations [51]. miR-451 may therefore have potential is a non-invasive diagnostic and prognostic biomarker for cancer. A reporter probe (Entry g, table 1) was then bound to the NPs using the ABisTES-8PEG conjugation. These nanoparticles were then used in a surface-based sandwich assay using the complementary capture probe and target miR-451 analogue (Entries e and f, table 1). This sandwich assay (figure 6(a)) was developed using surface chemistry and immobilization methods which reduces any NSB of the nanoparticles. We observed that the 8PEG-coated nanoparticles provide the least amount of NSB to our surfaces when compared to other nanoparticles we used. The capture probe was immobilized, followed by the introduction of the target miR-451 DNA analogue. After this, either the reporter probe-coated Cy5 NPs or a Cy5-conjugated reporter probe (Entry h, table 1) ('free dye') was added. Any NSB of target, reporter probes or nanoparticles was minimized by adding BSA to the surface after capture probe immobilization, followed by deactivating any remaining glutaraldehyde groups with sodium borohydride prior to addition of target and reporter probes. The Perkin Elmer Scanner fluorescent signal from the final reading of the Cy5 reporter and Cy5 nanoparticles bound to the probe areas is presented in figure 6(b). The limit of detection (LOD) of the two methods was calculated using the equations presented on the graph, which were determined by the fit of the two trendlines and from the standard deviation of the fluorescent signal at a target concentration of 0 pM. The minimum signal from the 0 pM sample shows that there is no NSB of the nanoparticles to the probe area. The calculated LOD for the free dye assay was 3.58 pM of target, whereas the LOD of the nanoparticle assay was 478 fM of target miR-451 DNA analogue, indicating that the use of the Cy5 high-brightness nanoparticles results in greater than a 7-fold increase in sensitivity than using the Cy5 dye alone.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. (a) Schematic outlining the process of the sandwich assay; (b) fluorescent signal measured for the sandwich assay using both reporter probe-coated Cy5-nanoparticles ('nanoparticle') and Cy5-labelled reporter probes ('free dye'). Error bars represent the standard deviations of three fluorescence intensity measurements. Equations used to calculate the limit of detection determined from the trendlines. Scanned at an instrument gain of 80.
Download figure:
Standard image High-resolution image{kind=link}
Conclusion
Oligonucleotide-conjugated Cy5-doped silica nanoparticles were prepared which showed excellent efficiency for the binding of DNA. The use of novel dipodal ABisTES functionalisation improved colloidal stability over monofunctional APTES-coated particles and, in combination with multivalent 8-arm PEG linkers, significantly improved DNA conjugation stability. We have shown that oligonucleotides bound to these NPs were available for hybridization to direct binding or sandwich binding targets in solution phase with no NSB. Finally, a DNA sandwich hybridization assay was carried out using these DNA-conjugated high-brightness nanoparticles as the reporter and LOD of less than 500 fM, a greater than 7-fold increase in sensitivity than using the Cy5 dye alone.
Acknowledgments
This material is based upon works supported by Enterprise Ireland and Science Foundation Ireland under Grants 10/CE/B1821, 14/TIDA/2334 and 14/TIDA/2369.
Supplementary information
Loss of DNA from APTES-coated nanoparticles.
Cy3 free dye calibration curve.
TEM images of functionalised nanoparticles.
Aggregation of nanoparticles over time.
Robustness of probe binding with no EDC/non-modified probes.
FRET investigations.